Chapter 12 LEfSe

We will use LEfSe (Linear discriminant analysis Effect Size) to determine which taxa can most likely explain the differences between the Western and Korean diet. LEfSe couples standard tests for statistical significance with additional tests encoding biological consistency and effect relevance. It can be used with other features such as organisms, clades, operational taxonomic units, genes, or functions.
In essence it allows for the detection of biomarkers when comparing sample groups. In the LEfSe terminology the sample groups are called the class.
Although LEfSe is a nice tool to use for a tutorial there are more robust tools.
Two recommended tools are:
They both require R and so are not included in this tutorial.
However, our R community analysis workshop does include how to use ANCOM-BC2.
12.1 Add metadata

We need to add metadata to our Bracken file to be ready for LEfSe. First we will copy the file so we have a backup in case we do anything wrong.
Using your favourite text editor (e.g. nano, vim, etc.) add the following line to the top of your all_num.lefse.bracken file. The words are separated by tabs. If you are not sure how to carry out this task please ask a demonstrator.
diet K K K K K K K K K K K K W W W W W W W W W W W W
Note: The above is diet followed by 12 K and 12 W.
The singular line should match the order of your samples within the file. This is the metadata line that LEfSe will use to determine which samples belong to each sample group, and therefore which to compare. In this case it is Korean diet samples versus Western diet samples.
12.2 Format file

We need to further format and preprocess our file with a LEfSe script.
Parameters

all_num.lefse.bracken: InputBrackenfile.all_num.lefse: Output file formatted for the run_lefse command, which we will soon run.-c 1: Specifies the row with the class info. This is used to determine which samples will be compared against which samples. In this case it is the first row with the Ks and Ws.-u 2: Specifies the row with the sample names. This is the second row in this case.-o 1000000: An integer can be indicated to determine to what size (count sum value) each sample should be normalised to.LEfSedevelopers recommend 1000000 (1 million) when very low values are present. We generally always use 1 million for consistency.
12.3 LEfSe run

Now to run LEfSe. All we need to do is run the command with the formatted input and provide an output file name.
12.4 Out file

The output file (all_num.lefse.out) is a tab-delimited file which contains a row for each species.
Biomarkers will have the five columns below whilst non-biomarkers will have the first two followed by a "-".
- Biomarker name
- Log of highest class average: I.e. get the class with the greater amounts of the biomarker, average the counts and then get the log of this value.
- Class with the greater amounts of biomarker
- LDA effect size: A statistical figure for
LEfSe. - p-value: Biomarkers must have a p-value of <0.05 to be considered significant.
The LDA effect size indicates how much of an effect each biomarker has. The default is to only count a species with an LDA effect size of greater than 2 or less than -2 as a biomarker. The further the LDA effect size is from 0 the greater the effect the species causes. Generally, it can be thought of as the order of magnitude difference in the abundance of the biomarker between the sample groups.
12.5 Biomarker LDA barchart
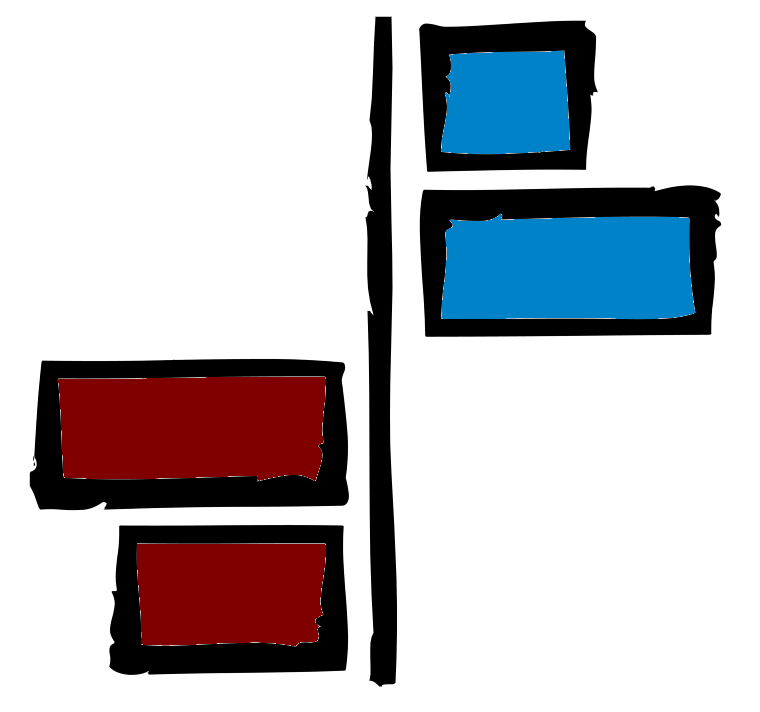
Next we can visualise the output.
--dpi 200: Dots per inch. This refers to the resolution of the output image. Normally publications want 300 dpi. We’ve chosen 200 as it is good quality and we will not be publishing these results.--format png: Format of output file. png is a commonly used file format for images.all_num.lefse.out:LEfSeoutput to visualise.biomarkers.png: Plot showing the LDA scores of the species detected as biomarkers. Colouring shows which class (K or W) the species is found in higher abundance.
Look at the figure with firefox:
12.6 MCQs

Interrogating the biomarkers.png plot and the all_num.lefse.out file, attempt the below MCQs.
Note: In this instance green bars represent biomarkers in higher abundance in the W samples whilst the red bars represent biomarkers in higher abundance in the K samples.
- Which species biomarker causes the biggest effect in the W class?
- Which species biomarker causes the biggest effect in the K class?
- Which species biomarker (i.e. present in the plot) causes the lowest effect in the W class?
- Which class has more biomarkers associated with it?
- What is the LEfSe p-value for Campylobactercoli?
- What is the Log of highest class average for Streptococcussuis?
- What is the LDA effect size for Bifidobacteriumlongum?
12.7 Kraken2 and Bracken databases
In your own future analysis you will need to create your own Kraken2 and Bracken databases. Please see the following links on information for this:
Kraken2- Standard
Kraken2databases: https://github.com/DerrickWood/kraken2/wiki/Manual#standard-kraken-2-database - Custom
Kraken2databases: https://github.com/DerrickWood/kraken2/wiki/Manual#custom-databases
- Standard
Bracken- https://ccb.jhu.edu/software/bracken/index.shtml?t=manual#step1
- This requires a
Kraken2database to be built first.