Chapter 4 GenBank and BLAST

GenBank and BLAST are both hosted by the NCBI (Nationl Center for Biotechnology Information). GenBank is an annotated database of all publicly available DNA sequences, more than 200 million. BLAST is a program that compares sequences to those in databases such as GenBank to find regions of similarity. Therefore we can use BLAST to find sequences similar to our ASVs in GenBank and use that to infer the taxonomy in our dataset.
This is not a fool-proof way to know exactly what was in our samples, however, as there are mis-annotated records in GenBank, which can occur if an organism is misidentified before being sequenced, or if the sequence was contaminated or erroneous. Although GenBank holds millions of sequences it is not comprehensive, and it might be the case that an ASV in our sample is simply not represented in the database.
It is possible to use BLAST through the web interface either pasting in sequences one by one or uploading a fasta file. However, with a few hundred ASVs from a typical metabarcoding project it is more efficient to use a downloaded copy of the GenBank nucleotide (nt) database and a standalone command line version of the BLAST software. See here for information on downloading BLAST software and databases. Many institutions will have servers with the BLAST software and databases communally available as this saves storage space compared to many users having their own copies, so it is worth asking if this is available where you are based.
As the BLAST databases are large and the searches can take a while to run the commands below are not intended to be run on the web VNC, but are provided in case they are useful for you for your own data.
BLAST is used on the linux command line, rather than in R as we have been working in all the earlier steps of the workbook.
4.1 BLAST againt the nt database
To perform a blastn search of our MiFish ASVs against the nt database the command would be:
blastn -query MiFish_ASVs.fa -task blastn -db nt \
-evalue 0.001 -outfmt 6 -out MiFish_ASVs_blast_out.txtThe options specified are:
-query filename.fafasta file of ASV sequences-task blastnperform a blastn search-db ntsearch against the nucleotide (nt) database-evalue 0.001threshold E-value for returning BLAST hits-outfmt 6generate the results in a tabular format
The text file produced will be a table of BLAST hits for all ASVs where a match could be found within the threshold E-value.
The table will have 12 columns including the ASV name, the database sequence match name, the percent identity of the match, the length of the alignment, the E-value and the bit-score. These last two values are measures of how well our sequences match, with a smaller E-value and a larger bit-score indicating better matches.
Here's what the first few lines of the output file looks like:
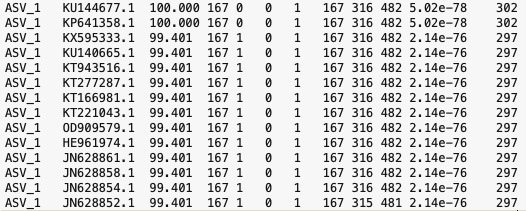
We have many hits for ASV_1. The first two hits have 100% sequence identity over the whole length (167 bp) of our amplicon. The other hits shown have 1 mismatch giving them a 99.4% sequence identity. How do we decide which match is best given the high similarity to many sequences? We could simply take the top hit for each ASV, but an alternative way is to use a 'lowest common ancestor' algorithm to assign taxonomy. This is implemented in the program taxonomizr.
4.2 BLAST against a custom database
If you have your own fasta file of curated reference sequences you can also use BLAST to match your ASVs to this.
First make a BLAST database from the fasta file:
This will generate all of the database files needed to run blastn against the sequences in 'My_12S_Fish_sequences.fa'. To do this the command would be: